










نگاهی به درمانهای ژنتیکی برای بیماری های زوال عصبی و مغزی
درمان بیماریهای زوال عصبی و مغزی یا نورودژرناتیو با رویکردهای ژنتیکی امیدوارکننده بوده است. برخی داروهای مبتنی بر AOS در مراحل بالینی قرار دارند.
وقتی سوزان برای نخستین بار به وجود چیزی غیرعادی درمورد مادرش پی برد هنوز کودک بود. اغلب اوقات هنگامی که مادرش قصد جابهجا کردن لیوان یا بشقابهای غذا را داشت، یا گاهی هنگام شستن ظرفها، آنها بهطور ناگهانی و بهظاهر تصادفی از دست مادرش میافتادند. سوزان چنین بهیاد میآورد که این اتفاقها برای مادرش شاید درنگاه اول صرفاً نشانهی بیدقتی یا حواسپرتی مادرش در آن لحظه میبود؛ اما او تأکید میکند که مادرش نمیتوانست آنگونه حواسپرت یا بیدقت باشد. درنهایت آنها بعد از مدتی متوجه میشوند که مادر از یک بیماری عصبی بهنام بیماری هانتینگتون یا HD رنج میبرد.
بیماری هانتینگتون عارضهای با زمینهی ارثی است که باعث ایجاد زوال گسترده در مغز انسان شده و مواردی ازقبیل فکر کردن، رفتارهای فرد، احساسات و حرکات فیزیکی او را دچار اختلال میکند. این بیماری بهطور معمول در سالهای میانی زندگی خود را نشان میدهد و اغلب با همین تغییرات جزیی در خلقوخوی فرد یا چالش در زمینهی تمرکز روی کارها آغاز میشود. با پیشرفت بیماری، فرد در ادامه دچار زوال عقل و ناتوانی در گفتار یا حرکت نیز میشود.
سوزان تنها یکی از افرادی است که فردی از خانوادهشان با بیماری HD مواجه شده است. اون از مجلهی ساینتیفیک امریکن درخواست کرده بود تا نام خانوادگی و هویتش بهمنظور حفظ حریم خصوصی، ناشناس باقی بماند. بااینحال موضوع مهم آن است که سوزان روزی که پی به بیماری مادرش برد را بهطور روشن و دقیق به یاد دارد. در بهار سال ۱۹۸۲ میلادی مادر را بهخاطر احساس خستگی بسیار شدید، افتادنهای مکرر و حرکات نامنظم به بیمارستان بردند. در آن زمان تستهای ژنتیکی خاصی برای تشخیص بیماری وجود نداشتند؛ درنتیجه پزشکان برای تشخیص بیماری دست به انجام تعدادی آزمایشها و ارزیابیهای مختلف زدند.
مقالهی مرتبط:پارکینسون؛ علائم، پیشگیری و درمان
معالج عصبشناس وی، تمام اعضای خانواده را به اتاقی فراخواند و بهطور مشخص درمورد وضعیت خاص بیمار با آنها صحبت کرده و برای آنها روشن کرد که بیماری درمانی نداشته و زمینهی ارثی هم دارد؛ و اینکه درصورت فرزندآوری سوزان و برادران و خواهرانش، امکان گسترش بیماری در خانواده به نسل بعد وجود خواهد داشت.
آن توضیحات ناگهانی و عجیب تأثیر عمیقی بر زندگی سوزان و برادرها و خواهرهایش گذاشت. برادرها تصمیم گرفتند که هیچگاه ازدواج نکنند. خواهر سوزان نیز تصمیم مشابهی گرفت و اقدام به عمل نابارورسازی کرد. اما برای سوزان اوضاع اندکی متفاوت بود؛ این گزینهها برای اون مفهومی نداشتند، چون او موقعی که با خبر بیماری مادرشان مواجه شد، باردار بود.
سوزان و همسرش در وهلهی نخست در تصمیم گرفتن برای انجام کار عقلانیتر ناتوان بودند. یکی از چالشهای فکری اصلی آنها این بود که اگر آنها کودک را به دنیا آورند، روزی در آینده همان کودک نیز با شرایطی مشابه شرایطی که آنها در آن قرار داشتند، مواجه خواهد شد. سوزان اشاره میکند که چنین شرایطی واقعا ظالمانه به نظر میرسید. درنهایت این زوج تصمیم ناراحتکننده و ناگزیر خودشان پیرامون پایان دادن به زندگی جنین را عملی کردند.
در ادامه درمورد بیماری هانتینگتون دقیقتر میشویم. ژن مرتبط با این بیماری با نام HTT شناخته میشود. این ژن، کدکنندهی پروتئینی بهنام هانتینگتین huntingtin است. نسخهی معیوب ژن، تکهای کوتاه از رشتهی خود را بهدفعات زیاد تکرار میکند. برخلاف برخی بیماریهای ژنتیکی که در آن برای پیدایش بیماری در بدن فرد نیاز به رخ دادن دو کپی معیوب از یک ژن وجود دارد، تنها ایجاد جهش در یک کپی از HTT کافی است تا فرد دچار بیماری هانتینگتون شود. ناقلان این جهش، بهمیزان ۵۰ درصد شانس انتقال آن به فرزندان خود را دارند. بهعبارتی، اگر فردی مبتلا به هانتیگتون باشد، بهاحتمال ۵۰ درصد فرزندش نیز در طی زندگی دچار جهش در این ژن و رخ دادن بیماری خواهد شد. سالها بعد از مرگ مادر سوزان، او و خواهر و برادرانش دریافتند که همگی این بیماری را از مادر خود به ارث بردهاند.
درحال حاضر هیچ درمانی برای متوقف یا کند ساختن پیشرفت این بیماری دردسترس نیست؛ این در حالی است که ما از سال ۱۹۹۳ میلادی با علت ژنتیکی آن بهطور آشکار و مشخص آشنا هستیم. بیشتر بیماریهای منجر به زوال مغز و اعصاب دیگر هم دارای درمانهای موثری نیستند؛ اگرچه سرچشمهی ژنتیکی آنها هم شاید نه بهاندازهی هانتینگتون، اما تا مقدار قابل توجهی برای دانشمندان روشن شده است و چندین دهه از شناسایی آنها میگذرد. از این قبیل بیماریها میتوانیم به ALS، آلزایمر و پارکینسون اشاره کنیم.
اکنون شاید اوضاع اندکی بتواند تغییر کند. بسیاری از پژوهشگران به داروهایی که تحت عنوان اولیگونوکلئوتیدهای آنتیسانس یا بهاختصار ASO شناخته میشوند، امیدوار هستند. ASOها بهزبان ساده، رشتههای کوتاهی از حروف DNA یا RNA هستند که بهمنظور متصل شدن به زنجیرههای خاصی از RNAهای ایجادشده توسط ژنهای معیوب طراحی شدهاند. به این طریق، سطوح پروتئینهای تولیدشده مجددا متعادل میشود. پروتئینهای ازدسترفته مجدد به مقدار نرمال خود برمیگردند و از سویی پروتئینهای معیوب و مازاد هم کاهش مییابند.
ادارهی غذا و داروی امریکا FDA در سال ۲۰۱۶ میلادی، اولین داروی ASO را برای یک بیماری عصبی تأیید کرد و از آن زمان تاکنون فعالیتهای چشمگیری در این حوزه انجام شده است. این زمینهی دارویی از جایگاه دو دههی پیش خود که صرفاً چند درمان بالینی محدود را شامل میشد، اکنون به حوزهای با روشهای درمانی زیاد برای انواع بیماریهای تخریبکنندهی اعصاب تبدیل شده و برخی از این درمانها هم به مراحل نهایی خود رسیدهاند.
مبتلایان به هانتینگتون بهمیزان ۵۰ درصد شانس انتقال آن به فرزندان خود را دارند
سایر پژوهشگران حوزهی داروهای ASO اکنون نگاه خود را به فراتر از بیماریهایی با یک جهش صرف، به بیماریهایی با شالودههای ژنتیکی پیچیدهتر متمرکز کردهاند. پیشرفتهای اخیر، بسیاری از افراد فعال در این حیطه را نسبت به آیندهی این فناوری خوشبین ساخته است. دان کلیولند، عصبشناس از دانشگاه کالیفرنیا سندیهگو، از نخستین دانشمندانی است که روی ASOها برای درمان بیماریهای عصبی تحقیق کرده است. او باور دارد که ما هنوز در اول راه هستیم و خبرهای بسیار بیشتری در آینده خواهیم شنید.
اما باید در نظر داشته باشیم که پیشرفتهای این عرصه بهآن اندازهای که تصور میرود، دارای مسیر هموار و یکنواختی نبوده است. در انتهای سال ۲۰۲۰، فاز سوم یکی از درمانهای مبتنی بر ASO بهخاطر عدم چربش مزایای دارو بر خطرها آن متوقف شد. از سویی برخی پژوهشگران همواره خواستار احتیاط پیرامون ASOها شدهاند. نگرانی اصلی آنها از این بابت است که ثمربخشی این درمانها در بسیاری از بیماریها هنوز نامشخص است؛ ضمن اینکه آنها معتقدند روش تزریق نخاعی این داروها هم مخرب و تهاجمی است.
با اینکه نتیجهی درمان آزمایشی اخیر ناامیدکننده بود اما کریس بوشف، مدیر پروژهی علمی ناظر بر درمانهای ژنتیکی در انستیتوی ملی بیماریهای عصبی و سکته امریکا معتقد است:
هنوز دلایل زیادی برای خوشبینی و اشتیاق نسبت به این روشهای درمانی و دستاوردهای آنها وجود دارد.
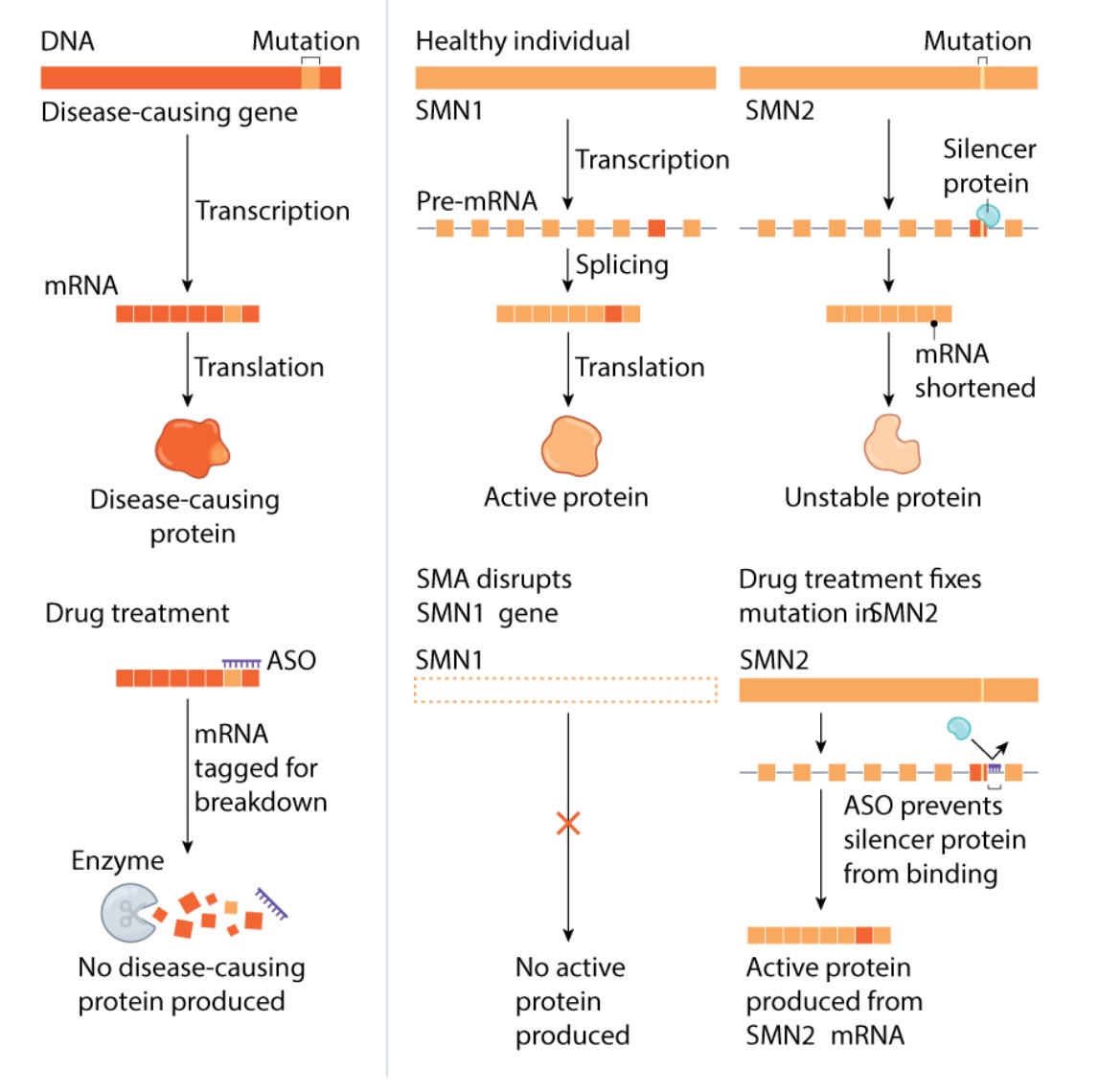
فرزند اول الیوت و ژانل لوئیس، در سال ۲۰۱۱ با یک بیماری نادر و ارثی زوال عصبی بهنام SMA یا تحلیل ماهیچهای ستون فقرات به دنیا آمد. افرادی مبتلا به SMA دارای نوعی جهش در ژنی موسوم به SMN1 هستند. این ژن در حالت عادی مسئول تولید نوعی پروتئین به نام SMN است. جهش در ژن یادشده باعث کمبود پروتئین SMN میشود و این کمبود باعث میشود تا مغز نتواند بهطور کارامد و طبیعی با بدن ارتباط برقرار کند. این ناتوانی در برقراری ارتباط به ضعف ماهیچهای و تحلیل رفتن میانجامد و در طی زمان هم بدتر میشود. چهار نوع مختلف از ژن SMA وجود دارد. متداولترین شکل آن SMA1 است که البته شدیدترین نوع هم محسوب میشود. افراد با جهش SMA1 بهطور معمول اندکی بعد از تولد علائم بیماری را بروز میدهند و بسیاری از آنها بیشتر از ۲ سال زنده نمیمانند.
بلیکلی، فرزند الیوت و ژانل، در سهماهگی علائم این بیماری را در خود نشان داد. الیوت اشاره میکند که در آن زمان درمانی وجود نداشت و بلیکلی در ۲۱ ماهگی جان خود را از دست داد.
این زوج در سال ۲۰۱۷، دختر دیگری به دنیا آوردند. اِوی، دختر دوم آنها هم مبتلا به SMA بود. اما او در قیاس با خواهر فقید خود، خوششانستر بود. چند ماه پیش از اینکه اوی به دنیا بیاید، FDA نوعی داروی ASO را تأیید کرده بود. این دارو بهنام نوسینرسین (nusinersen) شناخته شده و بهعنوان نخستین داروی دستکاریکنندهی سازوکار بیماری برای بیماری SMA بهشمار میرفت. اوی، اولین دوز از این دارو را در روز دوازدهم زندگیاش دریافت کرد.
دانشمندان برای نخستین بار در سال ۱۹۷۸ تواناییهای داروهای مبتنی بر ASOها را در هدف گرفتن RNA مورد بررسی و شناسایی قرار داده بودند؛ اما از آن زمان به بعد چندین دهه طول کشید تا آنها بتوانند به اطلاعات دقیقتری درمورد پتانسیلهای آنها برای درمانهای بالینی پی ببرند.
در اوایل مسیر، مشکلاتی چون سمی بودن و نداشتن توان درمانی پیشرفتها را کنند کرده و بهعنوان مانعی بر سر راه پروژهها مطرح شد. از همین رو، بسیاری از کمپانیهای داروسازی علاقهی اولیهی خودشان به این حوزه را از دست دادند و از ادامهی پژوهشها دست کشیدند. اما پژوهشگران فعال در یک شرکت داروسازی بهنام آیونیس Ionis در کالیفرنیا، توانستند با اعمال دستکاریها و تغییراتی بر شالودهی شیمیایی داروی اولیه، باعث افزایش توان درمان و پایداری دارو شوند و بدین ترتیب، داروهای ASO را به هدف مد نظر اولیه برسانند.
اشاره کردیم که داروی نوسینرسین نخستین داروی مورد تأیید FDA درنوع خود است. گامهای اولیه برای توسعهی آنچه که درنهایت به ساخت این دارو منتهی شد، حوالی سال ۲۰۰۰ میلادی در آزمایشگاه کولد اسپرینگ هاربر واقع در شهر نیویورک آغاز شده بود. در آن آزمایشگاه، یک متخصص ژنتیک مولکولی به نام آدریان کرینر مشغول تحقیق روی سازوکارهایی بود که به تولید پروتئینهایی با آسیبرسانی و پایداری کمتر توسط ژن SMN2 منتهی میشد. SMN2 هم یکی دیگر از ژنهای کدکنندهی پروتئین SMN محسوب میشود. آنها در پژوهشهای خود چنین استدلال میکردند که اگر بهطریقی بتوانند ژن SMN2 را وادار به تولید پروتئینهای بیشتری کنند، برای افرادی که دارای جهش در ژن SMN1 هستند ثمربخش و جبرانکننده خواهد بود. آنها با بررسی کارهای سایر گروههای پژوهشی پی برده بودند که تقریباً در همهی بیماران، علت ایجاد مشکل با ژن SMN2، رخ دادن یک خطا در طی فرایند اتصال است. فرایند اتصال به روندی گفته میشود که در آن رشتههای RNA برش زده شده و وارد پروسهای از دستورالعملهای ساخت پروتئین میشوند. این فرایند باعث میشود تا تکهی از کد ژن SMN2 محفوظ باقی بماند.
نوسینرسین زندگی بسیاری از کودکانی را که بیشتر از ۲ سال زنده نمیماندند، نجات داد
گروه کرینر روی پروتئینهایی تمرکز کردند که به رشتههای RNA پیوند خورده و موجب محفوظ ماندن همان تکهی یادشده میشدند. آنها امیدوار بودند که بتوانند بهطریقی مانع دخالت این تکهها در فرایند تولید پروتئینهای کامل SMN شوند. در سال ۲۰۰۴، کرینر شروع به همکاری با فرانک بِنِت، داروشناس و یکی از اعضای پایهگذار کمپانی آیونیس کرد. آنها با همکاری یکدیگر درنهایت موفق به شناسایی دقیق نوعی ASO شدند که قادر به چسبیدن به رشتههای RNA و پنهان ساختن بخش مورد نظر آن رشته، از پروتئینهایی بود که تمایل به از کار انداختن آنها داشتند. بدین ترتیب، زمینه برای تولید پروتئینهای SMN کارامد و سالم فراهم میشد.
این ماده که با نام نوسینرسین شناخته میشود، در سال ۲۰۱۱ وارد مرحلهی درمانهای بالینی آزمایشی شد. نتایج درمانهای آزمایشی بهاندازهای امیدوارکننده بودند که فاز سوم آزمایش دارو در نوزادان مبتلا به بیماری SMA بسیار زود پایان یافت. نتیجهی نهایی این بود که احتمال ادامهی حیات و گذر از حد نصاب ۲ سالهی مد نظر در این نوزادان، در بیمارانی که این دارو را دریافت کرده بودند بسیار بیشتر بود.
تا این زمان، بیش از ۱۰ هزار نفر در سطح جهان داروی نوسینرسین (با نام تجاری Spinraza) را دریافت کردهاند. دارو بهطور چشمگیری مسیر متداول و پیشفرض این بیماری کشنده را تغییر داده است. کودکانی که به SMA مبتلا هستند و اندکی پس از تولدشان این دارو را دریافت میکنند، دیگر به روال سابق در سالهای اول زندگیشان نمیمیرند.
راسل باترفیلد، عصبشناس اطفال در دانشگاه یوتا که بهعنوان مشاور در این پروژهها فعال بوده است، اشاره میکند که مکالمهاش با والدین این نوزادان به ابراز ناامیدی صرف منتهی نمیشود. باترفیلد میگوید در حال حاضر آنها با امیدواری میتوانند به والدین کودکان بگویند که داروی جدیدی برای این بیماری نادر وجود دارد که عملکرد شگفتانگیزی هم داشته است و هرچه سریعتر باید کارهای درمان را آغاز کنند.
اِوی لوئیس هماکنون ۴ سال دارد و هر چند ماه یک بار، یک دوز از داروی اسپینارازا را ازطریق تزریق در ناحیه کمر دریافت میکند و تاکنون ۱۵ بار این دارو را دریافت کرده است. بااینکه اون هنوز هم برخی مشکلات از قبیل تغذیه ازطریق تیوب مخصوص دارد، ولی میتواند راه برود، بدود و بازی کند؛ چیزهایی که خواهر اون بلیکلی هیچگاه قادر به انجامشان نبود و در اثر همین بیماری هم جان خود را از دست داد.

اوی لوئیس ۴ ساله که بهخاطر درمانهای جدید هانتیگتون زنده ماند
برنامه ای جامع برای سایر بیماری های مشابه
در پی موفقیت نوسینرسین، پژوهشگران شروع به بررسی امکان درمان سایر بیماریهای با منشا جهش ژنی مشخص، از قبیل بیماری هانتینگتون کردند. این تلاشهای درادامه به تولید داروی تومینرسن tominersen انجامید. این دارو نیز توسط شرکت آیونیس توسعه داده شده بود و مجوز آزمایشهای بالینی آن هم به کمپانی دارویی روشه در بازل سوئیس اعطا شد.
تصور میشود که داروی فوق با هدف قرار دادن تکرارهای CAG روی رشته RNA تولیدشده توسط ژنهای HTT طبیعی و معیوب، و برچسب زدن آنها برای تخریب توسط آنزیمی به نام RNase H1 کار کند. نتایج کارآزمایی بالینی فاز اول و دوم که در سال ۲۰۱۹ منتشر شد، نشان داد که تومینرسن غلظت نسخهی جهشیافتهی هانتینگتین را در مایع مغزی نخاعی، بدون ایجاد عوارض جانبی جدی، کاهش داده است.
مقالهی مرتبط:بیماری آلزایمر : هر آنچه باید درباره زوال عقل و نشانه ها و علائم آن بدانیم
موفقیت آزمایش اولیهی هانتینگتون توجه محققان تخریب عصبی را به خود جلب کرد، زیرا درهمتنیدگی پروتئین یکی از ویژگیهای کلیدی در بسیاری از اختلالات مغزی و عصبی است. سارا تبریزی، متخصص مغز و اعصاب کالج لندن میگوید:
هیجان زیادی در مورد این موضوع وجود داشت؛ زیرا این موفقیت اولیه واقعاً درهایی را باز کرد تا بتوانیم آزمایشهای بالینی ضدحسی را برای سایر بیماریهای تخریبکنندهی عصبی انجام دهیم؛ بیماریهایی که در آن، ایجاد یک پروتئین جهشیافتهی سمی نقش اساسی دارد.
گفتنی است که تبریزی، مرحلهی اول و دوم آزمایش بالینی تومینرسن را هدایت کرده است.
اما یک خبر غیرمنتظره در پایان ماه مارس ۲۰۲۱ (بهار ۱۴۰۰) شوک بزرگی به جامعهی بیماران هانتینگتون و دانشمندان فعال در زمینهی آن وارد کرد. آزمایش فاز سوم تومینرسن که شامل ۷۹۱ شرکتکننده از ۱۸ کشور جهان بود، به توصیهی یک کمیتهی مستقل از کارشناسانی که بررسی برنامهریزیشدهای روی دادههای آزمایش انجام داده بودند، زودتر از موعد به پایان رسید. در بیانیهای از کمیتهی روشه چنین آمده بود:
هیچ نگرانی ایمنی جدیدی ظاهر نشده است؛ اما مزایای بالقوهی دارو هم بر خطرهای احتمالی آن چربش نداشت.
تبریزی میگوید تا زمانی که جزئیات بیشتری منتشر نشود، نمیتوان بهطور دقیق گفت که چه اشتباهی رخ داده است.
البته نباید از یاد ببریم داروهایی که با روشی مشابه تومینرسن عمل میکنند، هنوز برای سایر اختلالات مغزی و عصبی با علتهای مشابه، روی میز هستند. بهعنوان مثال، برخی از موارد بیماری عصبی ALS بهدلیل وجود بیش از حد یک پروتئین جهشیافته ایجاد میشود و تعداد انگشتشماری از داروهای مبتنی بر ASO برای همان شکلهای خاص از این بیماری در مرحلهی آزمایشهای بالینی قرار دارند. از میان آنها توفرسن بیشترین مسیر را در جهت رسیدن به نتیجهی نهایی پیموده است. توفرسن داروی مبتنی بر ASO است که توسط آیونیس برای درمان گونهی ارثی از بیماری ALS توسعه یافته است. توفرسن اکنون در مرحله کارآزمایی فاز سوم تحت حمایت شرکت بیوژن در حال طی آزمایشهای مربوطه است.
کلودیا تستا، متخصص مغز و اعصاب در دانشگاه ویرجینیا کامنولث در ریچموند، اشاره میکند که کاهش سطح پروتئین جهشیافته، مانند کاری که با داروهای تومینرسن و توفرسن انجام میشود، در مقایسه با تقویت پروتئین از دسترفته، مانند آنچه که نوسینرسن صورت میدهد، چالشهای منحصربهفردی دارد. شماری از استراتژیهای کاهش پروتئین، درواقع سطوح نسخههای خوب و بد یک پروتئین را کاهش میدهند. دانشمندان هنوز از اثرات بلندمدت آن بر بیماریهای مربوطه اطلاعی ندارند و مشخص نیست که آیا این موضوع در مرحلهی سوم آزمایش تومینرسن مد نظر بررسیکنندگان بوده است یا خیر. تستا میگوید:
داروی SMA اساساً کار متفاوتی انجام میدهد؛ از همین روی، نمیتوان بر مبنای آن، اثربخشی دارو برای سایر بیماریها را پیشبینی کرد و این یک حقیقت دردناک است.
برخی از داروهای مبتنی بر ASO برای جلوگیری از این دست مسائلی که بالاتر اشاره کردیم، پروتئینهای جهشیافته را بهطور دقیق و مشخص هدف قرار میدهند. یک شرکت بیوتکنولوژی به نام Wave Life Sciences در کمبریج ماساچوست، در حال آزمایش نوعی استراتژی است. آنها در استراتژی خود جهشهای کوچکی را که گاهی در کنار تکرارهای CAG و تنها روی نسخههای جهشیافتهی HTT رخ میدهند، هدف قرار میدهد. هدف این است که سطح هانتینگتین سالم نسبتاً دستنخورده باقی بماند. اما این دارو فقط در زیرمجموعهای از افراد مبتلا به هانتینگتون که حامل این جهش ها هستند کار میکند. بهگفتهی تستا، تفاوت موجود در این سطح را تنها با روش توالییابی جامع و دقیقی که بهطور معمول در کلینیکها انجام نمیشود، میتوان شناسایی کرد.
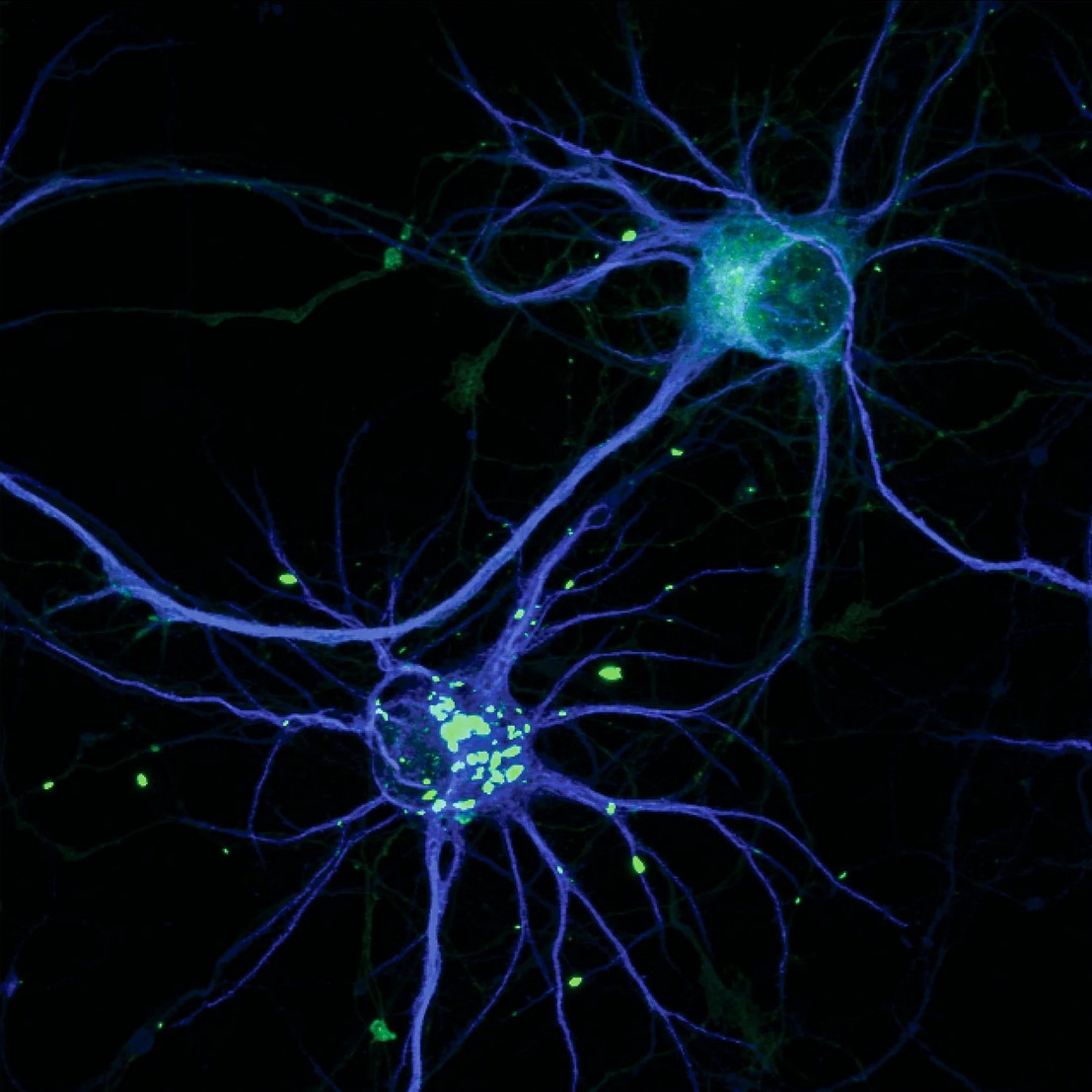
نسخهای سمی از پروتئین HTT عامل بیماری هانتینگتون منجر به ایجاد تودههایی میشود که با رنگ سبز روشن روی تصویر مشهود است
محققان بهتازگی شروع به آزمایش درمانهای مبتنی بر ASO برای بیماریهای تخریبکنندهی عصبی شایعتری مانند پارکینسون و آلزایمر کردهاند. اکثریت قریب به اتفاق موارد بیماریهای یادشده، به یک جهش ژنتیکی خاص مرتبط نیستند و این اختلالات بسیار بیشتر از بیماریهای ارثی کمیابتری هستند که در بخشهای بالاتر این مقاله به آنها پرداختیم. هدف داروهای مبتنی بر ASO در مواجهه با بیماری آلزایمر، کاهش سطوح پروتئینی بهنام تاو است. تاو پروتئینی است که بهصورت بخشهای درهمتنیدهی سمی در مغز شکل میگیرد. همچنین در بیماری پارکینسون، هدف دارو بر کاهش پروتئینی موسوم به آلفاسینوکلئین است؛ پروتئینی که در تودههای پاتولوژیکی بهنام اجسام لویی تجمع مییابد.
کوین تالبوت، متخصص مغز و اعصاب دانشگاه آکسفورد بریتانیا، که در آزمایش آتی داروهای مبتنی بر ASO برای درمان بیماری ALS شرکت خواهد کرد، میگوید:
… بااینحال برای بیماریهای عصبی مانند اینها، احتمالاً چندین ژن در یک شبکه درگیر هستند. مشخص نیست که تغییر یک ژن در شبکه چگونه روی بقیه تأثیر میگذارد.
تالبوت پیشتر در هیئتهای مشاورهی علمی روشه و بیوژن فعالیت کرده است.
بهگفتهی تالبوت موضوع دیگری که باید توجه کنیم این است که داروهای فوق در حال حاضر باید ازطریق ایجاد سوراخهای کمری مکرر برای رسیدن به اهداف مد نظر به سیستم عصبی مرکزی تحویل داده شوند. تالبوت اشاره میکند:
پیش از به کار برده شدن داروهای مبتنی بر ASO در طیف وسیعتری از بیماریها، یافتن راهی برای عبور دادن این داروها از سد خونی-مغزی بهمنظور انتقال آنها بهصورت کمتر تهاجمی، بسیار مهم خواهد بود. فهرستی کامل و مفصل از کارهایی وجود دارد که باید پیش از پرداختن به جشن پیروزی نهایی، انجام دهیم.
تغییر هویت
مطالعات روی موشها نشان میدهد که داروهای ASO در آینده میتوانند کاربردهای قدرتمندتری در مغز انسان داشته باشد: جایگزینی نورونهای از دست رفته.
سال گذشته، شیانگ دونگ فو، زیستشناس سلولی و همکارانش در دانشگاه سندیهگو کالیفرنیا، نشان دادند که میتوان از ASO برای تبدیل سلولهای غیرعصبی مغز به نام آستروسیتها به نورون استفاده کرد. این تیم یک داروی مبتنی بر ASO را به ناحیهای از مغز موش که نورونهایش در طی بیماری پارکینسون از بین میروند، تزریق کردند. زمانی که دارو به محل مورد نظر رسید، شبکهای از ژنها را فعال کرد؛ ژنهایی که آستروسیتها را وادار به تبدیل شدن به نورونهای جدید میکرد. گروه پژوهشی فو در مدلهای بررسیشده در بیماری پارکینسون مغز موش، به این نتیجه رسیدند که حیوانات دریافتکنندهی این درمان، در رفتارهای خاصی بهبودی نسبی از خود نشان دادهاند.
دان کلیولند، در آزمایشهای بالینی فو شرکت داشته و با یک داروی مبتنی بر ASO ارائهشده توسط ایونیس برای آزمایش این ایده در قسمتهای دیگر مغز کار کرده است. او میگوید:
اینجا واقعا همان حوزهای است که میخواهم باقی دوران کاری خود را روی آن سرمایهگذاری کنم. مطمئن هستم که ما فقط در آغاز مسیری طولانی درمورد احتمالات موجود هستیم.
داروهای ASO تبدیلکنندهی آستروسیت هنوز در مراحل اولیه هستند. فو هشدار میدهد که این تکنیک قبل از انتقال به مرحلهی بالینی، باید روی پستانداران غیرانسان آزمایش شود؛ زیرا مطابقت مغز آنها با مغز ما بیشتر از مطابقت مغز موشها با مغز انسان است.
در حال حاضر، محققان مشتاقانه منتظر نتایج آزمایشهای فاز سوم توفرسن برای بیماری ALS هستند. آنها میخواهند دقیقاً بدانند که چرا آزمایش تومینرسن برای هانتینگتون متوقف شد.
سوزان، پرستار بازنشستهای که در اواسط دههی هفتم زندگی خود به سر میبرد و از مرحلهی اول در آزمایشهای تومینرسن شرکت داشته است. او میگوید که از شنیدن خبر توقف زودتر از موعد آزمایشهای بالینی فاز سوم ناامید شده است؛ اما از توجه و اهمیتی که بهعنوان یک شرکتکننده دریافت کرده خشنود است.
من از روز اول این افتخار را داشتم که عضوی از این آزمایشها باشم. اکنون راهی جز صبر کردن و بررسیهای بیشتر نداریم. هیچ راه جایگزینی وجود ندارد.